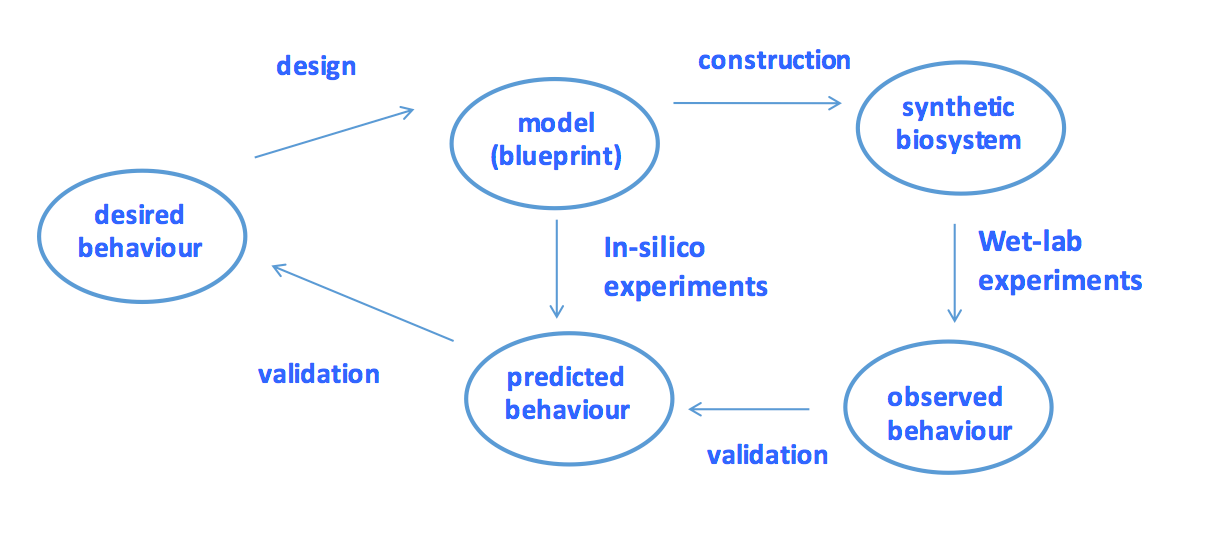
Computational, mathematical and statistical approaches play vital roles in the design and analysis stages of the synthetic biology workflow. This is illustrated in the figure below, where desired behaviour is encoded as a functional phenotype and described by a mathematical model which acts as a blueprint or design template to guide the construction of the physical biological system. These models need to be analysed using advanced mathematical and computational techniques for internal consistency and coherence, as well as checking that they properly capture the properties of the desired target behaviour. Moreover, the actual behaviour of the implemented biosystems needs to be checked against the desired behaviour as expressed by the design templates, and against the desired target behaviour. As with any engineering process, the workflow is best conceived of as a set of cycles, representing refinement of the design and the implementation.
The source underlying data for the design templates is the genomic information which has been generated through the sequencing activities of the Microbial BioEngineering cluster. These data are large because they describe the genomic content of several bacterial micro-organisms each with many distinct strains, and contain the variability inherent in living systems which are subject to mutations. Thus statistical analysis and related machine learning approaches are vital components of our analysis and design methodology.
Our activities currently focus on computational design. Other areas of expertise include: statistics, machine learning, evolutionary computation, optimisation, artificial intelligence, biomedical informatics, data science.
Level 2: Computational design of biological systems (Leader: David Gilbert)
The Synthetic Biology Computational Design Group focusses on the development and application of methodologies for the design of novel microbacterial strains, in collaboration with the Microbial BioEngineering Group. This research involves the use of models at the levels of biochemical networks and also of the structures of their constituent proteins as design ‘templates’ which can act as guidelines for bioengineering implementation. These two complementary modelling approaches are combined in powerful multilevel and thus multiscale models. This will be achieved using computational analysis to engineer design solutions over multiscale levels from polymorphism at the genetic level and its effect at the protein structure and biochemical pathway levels. The Group members carry out the following areas of activities:
Level 3: Network model based design
Keywords: network/model modification, graph editing, network/model analysis, model checking, multiscale modelling, databases
Network models describe the metabolic biochemical reactions of bacterial strains as systems of continuous or stochastic equations, resulting in an overall bipartite graph structure which can be represented as a Petri net. Our work involves the activities of model construction, analysis and modification. The models can be analysed for both their static and dynamic properties in order to confirm that they are sound and consistent, and checked against observations of the bacteria that they describe.
We are developing a modelling database facility to store components from public domain models as well as locally generated data which can be reused for model construction. The database will include phenotypic annotation and direct links to public molecule and reaction databases. Integration with the database of the Brunel Strain Collection will facilitate implementation of the process from in silico design to in vitro / in vivo experiments. The information stored will be extended from gene data to include proteins and metabolites.
The generation of designs for new synthetic bacterial strains involves the selection of optimal combinations of chassis (host strain) and genes for transfer, knockout or modification. This is achieved using selection, mutation and optimisation techniques on the computational models, resulting in models of virtual organisms which can be used as in-silico designs to guide the construction of novel strains.
Level 3: Protein structure informed design
Keywords: protein structure, protein function, protein-protein interaction networks
Metabolic biochemical reactions are performed by proteins. Knowledge of their 3D structure has been proven critical to understand the molecular effect of variations in the amino acid sequence on their catalytic activity, and thus desired phenotypic changes are often delivered through modification in protein function. The effect of functional polymorphisms will be modelled at the protein structure level for the key players in metabolic pathways. The effect of mutations on the dynamics and kinetics of the protein will be modelled to inform design strategies. Ligand and protein-protein interactions will be simulated and integrated with biochemical modelling using a multiscale strategy.
We aim at providing a set of computational tools for the selection of mutations for adaptive improvement of E. coli to confer susceptibility to selected antibiotics. The tool will comprise a database of naturally evolved mutations from E. coli strains (provided by the MBE cluster) mapped on the bacterial 3D protein-protein interaction network (PPIN) and a predictive software to mine the database for candidate mutations with potential to confer adaptive and desired phenotypes when combined.
We are developing a database protein structures, interactions and mutations. The most recent and comprehensive PPIN for E. coli has been used as reference. Experimental structures have been integrated with theoretical models.
The core genome from Brunel E. coli strain collection has been mapped to the PPIN and the unique set of 3D proteins structures for the core genome have been used to populate a relational database. A mapping of non-synonymous mutations in coding regions from naturally evolved E. coli strains is in progress. Each protein entry will contain information on mutations, associated strains and experimental conditions.
Structural models of proteins with selected mutations will form a library of design templates that will be integrated into multilevel biochemical network models with desired phenotypic behaviours.